
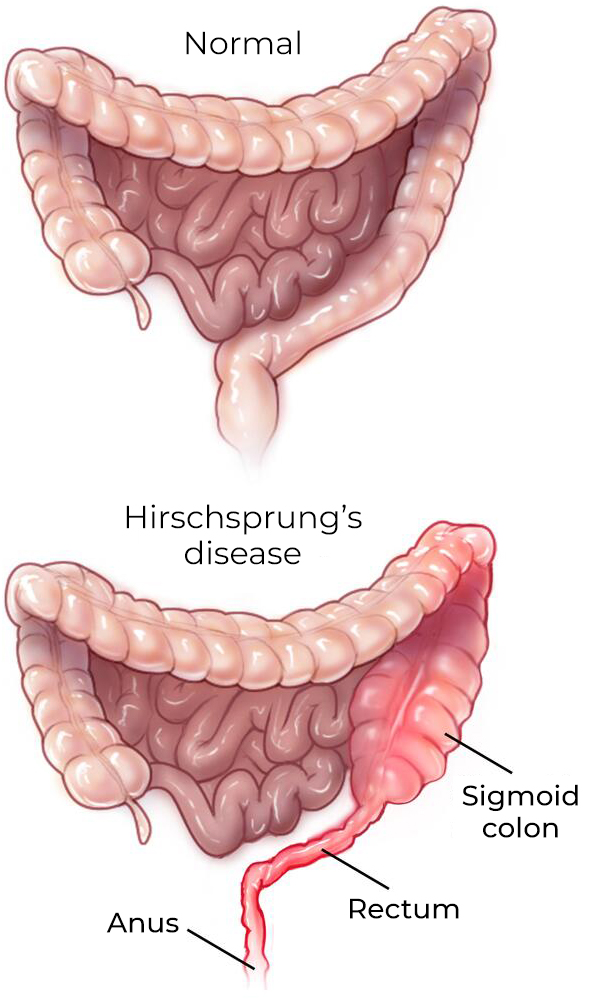
Hirschsprung disease, named after the Danish pediatrician Harald Hirschsprung (1830-1916), is a rare congenital anomaly of the enteric nervous system. It is characterized by the absence of ganglion nerve cells in the terminal part of the rectum, which causes functional intestinal obstruction in the newborn. It is diagnosed in approximately 80% of patients during the neonatal period, that is, during the first four weeks of life.
The disease is classified into two types based on the length of the aganglionic intestinal segment:
- SHORTSEGMENT
- The most common form, where the diseased segment encompasses only the rectosigmoid colon.
- LONGSEGMENT
- The aganglionic segment extends beyond the sigmoid colon and in some cases reaches the junction with the ascending colon.

HIRSCHSPRUNG DISEASE
Hirschsprung disease results from a defect in the migration of neural crest cells during embryonic development. The migration of neural crest cells to the digestive tract begins in the 4th week and is completed by the 7th week of gestation. A defect in the development of the enteric nervous system leads to incomplete colonization of neuronal and glial cells in the colon, causing the varying lengths of aganglionic segment observed in Hirschsprung disease.
Since the enteric nervous system is responsible for a wide range of critical gastrointestinal functions, the absence of enteric ganglia in the distal colon results in severe constipation (megacolon), enterocolitis (inflammation), bacterial translocation into the bloodstream, sepsis, and premature death in patients with Hirschsprung disease.
Epidemiology and Genetics of Hirschsprung Disease
The global incidence of Hirschsprung disease is 1 to 2.6 cases per 10,000 births. The condition is more common in males than females, with a male-to-female ratio ranging from 2.8:1 to 4:1 for the short-segment form.
Approximately 80% of Hirschsprung disease cases are sporadic, but the condition has also been shown to be associated with other genetic or chromosomal abnormalities such as Down syndrome. In fact, 10% of Hirschsprung disease cases occur in children with Down syndrome. The genetics of Hirschsprung disease are highly complex, involving more than 20 genes. Genetic aberrations most associated with the disease however involve the RET gene, which encodes a transmembrane tyrosine kinase activated by GDNF when bound to co-receptor GFRα1. Indeed, about 50% of children with the short-segment form of Hirschsprung disease carry non-coding variants of the RET gene resulting in reduced, but not absent, RET activation.
TREATMENT AND COMPLICATIONS
The only treatment currently available for Hirschsprung disease is surgical removal of the aganglionic segment of the colon followed by reconnection of the innervated colon to the anus. This procedure is painful and costly, and unfortunately, postoperative complications such as fecal incontinence and potentially life-threatening inflammation (enterocolitis) are common, often leading to hospital readmission and additional surgeries. Moreover, gastrointestinal issues frequently persist into childhood and may continue into adulthood, affecting quality of life. Post-surgery, nutritional support is essential to ensure adequate growth and development of children with Hirschsprung disease, and regular monitoring of bowel function and overall health is required.

